

The FDA has introduced new regulations for wearable medical devices, transitioning from the Quality System Regulation (QSR) to the Quality Management System Regulation (QMSR) on February 2, 2026. This update aligns with the global ISO 13485:2016 standard, creating stricter rules for the design, production, and testing of wearable devices in the U.S.
Here’s what you need to know:
- What Changed? QMSR replaces QSR, focusing on risk-based processes and international alignment. It impacts everything from design to post-market inspections.
- Who’s Affected? Manufacturers of Class II and III wearables, especially those with software or AI components.
- Key Requirements:
- Design Controls: Devices must meet user needs and pass real-world testing.
- Process Controls: Manufacturing processes must ensure consistent quality, especially for software and critical components.
- Testing Protocols: Biocompatibility, software validation, and risk analysis are mandatory.
- Non-compliance Risks: Devices may be deemed "adulterated", leading to FDA actions and market restrictions.
The shift to QMSR raises the bar for wearable device testing, requiring manufacturers to adopt detailed risk management and validation processes. Following these steps ensures compliance and safe, reliable devices for consumers.
QSR to QMSR: The Rewrite of 21 CFR Part 820 & Key Considerations for FDA Compliance
sbb-itb-44aa802
FDA QSR Requirements for Wearable Devices
The Quality Management System Regulation (QMSR) outlines three core principles for developing, manufacturing, and testing wearable devices. These rules apply to Class II and Class III wearables and even Class I devices equipped with automated software. Each principle addresses a distinct phase of the device lifecycle, covering everything from the initial concept to final distribution.
Design Controls and Validation
Under 21 CFR 820.30 (aligned with ISO 13485 Clause 7.3), manufacturers must follow a structured process to turn user needs into verified and validated products. For wearables, this starts by defining key specifications like sensor accuracy, battery life, and skin compatibility.
Verification ensures that the design outputs meet the initial inputs. For instance, if a wearable’s heart rate sensor is expected to maintain an accuracy of ±5 beats per minute, verification tests must confirm this under controlled conditions. Validation takes this further by testing early production units in real or simulated use scenarios. A device might pass lab verification but fail validation if practical issues – like unclear labeling or discomfort during use – arise.
Risk analysis plays a critical role throughout the design process. Manufacturers need to assess hazards under both normal and fault conditions, such as sensor malfunctions, overheating batteries, or data errors.
"Design validation shall be performed under defined operating conditions on initial production units, lots, or batches, or their equivalents." – 21 CFR 820.30(g)
Once the design is validated, strict production and process controls ensure that the manufacturing process consistently produces devices that align with the validated design.
Production and Process Controls
Production and process controls, as outlined in 21 CFR 820.70, are essential for ensuring wearables are manufactured reliably. These controls are especially important for processes that can’t be fully verified later, such as assembling sensors, sealing waterproof components, or bonding adhesives. In these cases, process validation (required by 21 CFR 820.75) ensures that every production run meets quality standards.
Other safeguards include environmental monitoring to maintain optimal temperature and humidity, thorough training for staff, and routine equipment maintenance to prevent defects. For wearables with high-density lithium batteries, these measures are critical to avoid safety risks. Additionally, software and firmware must meet the same quality standards as physical components. Any updates to code or algorithms must adhere to documented change control procedures.
After production, devices undergo multiple layers of inspection and testing to ensure they meet all quality and safety benchmarks.
Inspection and Testing Protocols
The FDA enforces a three-stage inspection and testing process to prioritize patient safety. These protocols, detailed in 21 CFR 820.80-86, include:
- Receiving inspection: Ensuring incoming components like sensors, batteries, and circuit boards meet the required specifications.
- In-process inspection: Monitoring assembly stages to catch potential issues early.
- Final device acceptance: Verifying that finished devices meet all performance, safety, and labeling requirements before distribution.
Testing for wearables is tailored to their unique features. For example, biocompatibility tests (following ISO 10993) confirm that materials in contact with skin won’t cause irritation, while electromagnetic compatibility tests (per IEC 60601-1-2) ensure wireless functions don’t interfere with other medical devices. Environmental testing subjects devices to conditions like humidity, shock, and vibration to simulate real-world use.
The FDA’s Compliance Program 7382.850, introduced on February 2, 2026, replaces the Quality System Inspection Technique (QSIT). This program allows inspectors to review internal audits, supplier evaluations, and management reports. As a result, manufacturers must maintain detailed records of all testing, including servicing activities and investigations into device failures.
| Requirement Area | Relevant Regulation/Standard | Testing Impact |
|---|---|---|
| Design Verification | ISO 13485 Clause 7.3.6 | Confirms sensor and battery outputs meet technical specifications. |
| Design Validation | ISO 13485 Clause 7.3.7 | Tests under actual or simulated use conditions to ensure the device meets user needs. |
| Process Validation | 21 CFR 820.75 | Ensures consistent performance in processes that cannot be non-destructively tested. |
| Software Validation | 21 CFR 820.30(g) | Verifies firmware performance and mobile app functionality. |
| Risk Analysis | ISO 13485 Clause 7.1 | Directs testing to address specific hazards like skin irritation or data loss. |
How QSR Affects Wearable Device Risk Classification
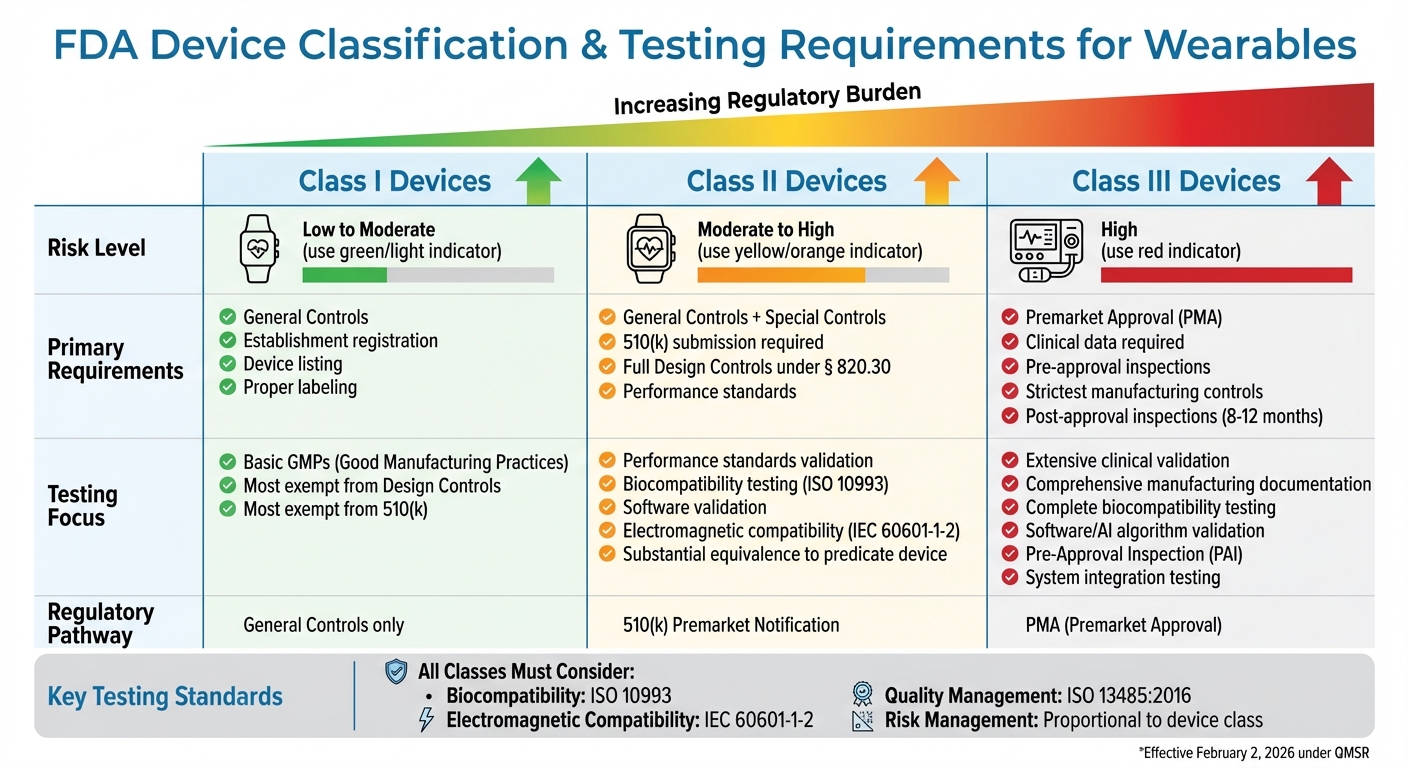
FDA Device Classification and Testing Requirements for Wearables
The FDA’s risk classification system plays a key role in determining the level of testing and documentation a wearable device needs. Devices are grouped into three classes based on the potential risk they pose to users. As the class number increases from I to III, the regulatory requirements become progressively stricter. This framework ensures that testing protocols align with the Quality Management System Regulation (QMSR).
Class I devices are considered low to moderate risk and are subject to "General Controls." These include basic requirements like registering the establishment, listing the device, and ensuring proper labeling. Most Class I devices only need to follow basic Good Manufacturing Practices (GMPs) and are generally exempt from full Design Controls unless explicitly required by their classification.
Class II devices fall into the moderate-to-high risk category. In addition to General Controls, they must meet "Special Controls", which might include performance standards, postmarket surveillance, or specific labeling requirements. These devices typically require a 510(k) submission and must comply with full Design Controls under § 820.30 to confirm they perform as intended.
Class III devices pose the highest risk, often being life-supporting or life-sustaining. These devices require Premarket Approval (PMA), which involves clinical data, pre-approval inspections, and the strictest manufacturing controls.
The classification of a wearable device heavily depends on its "intended use" and "indications for use", as outlined in its labeling or marketing claims. For instance, a wearable marketed for general wellness without medical claims typically avoids FDA regulation. However, if it includes claims like "detects atrial fibrillation", it may be classified as a Class II or III device, triggering full QMSR requirements.
Testing Requirements for Class II and Class III Wearables
Testing protocols become more rigorous as the device’s risk level increases. Class II devices must prove they are substantially equivalent to a legally marketed predicate device through the 510(k) process. Testing focuses on meeting performance standards and special controls specified in their classification. For example, a Class II heart rate monitor needs to validate sensor accuracy under various conditions and demonstrate reliability in real-world scenarios.
Class III devices, on the other hand, require extensive clinical validation, comprehensive manufacturing documentation, and a Preapproval Inspection (PAI) before receiving FDA approval. Once approved, the FDA may conduct post-approval inspections within 8 to 12 months, focusing on any changes in the device’s design, manufacturing, or quality systems.
For wearable devices powered by AI, testing must address potential safety risks. This includes detailed software validation and system integration testing, with the depth of testing proportional to the device’s risk level. Manufacturers must also ensure that supplier selection, software validation, and corrective actions align with these risk considerations.
Testing for all device classes often includes biocompatibility (per ISO 10993) and electromagnetic compatibility (per IEC 60601-1-2) to ensure both safety and functional accuracy.
| Device Class | Risk Level | Primary Requirement | Testing Focus |
|---|---|---|---|
| Class I | Low to Moderate | General Controls | Basic GMPs; most exempt from Design Controls and 510(k) |
| Class II | Moderate to High | Special Controls + 510(k) | Performance standards, biocompatibility, software validation |
| Class III | High | Premarket Approval (PMA) | Clinical validation, pre-approval inspections, complete manufacturing documentation |
Examples of QSR-Compliant Wearables
The aiSpine posture monitoring device by AIH LLC provides a practical example of how a wearable can meet QSR requirements for medical device classification. Designed to support spinal health management, aiSpine adheres to Design Controls under § 820.30, maintaining a Design History File that documents risk analysis, sensor accuracy testing, and validation under real-world conditions. The device’s integration with the AIH Health App for real-time feedback necessitates software validation tailored to its risk level.
Similarly, the aiRing vital signs monitoring ring showcases QSR compliance. This wearable tracks multiple physiological parameters and must meet biocompatibility standards for prolonged skin contact, electromagnetic compatibility for wireless data transmission, and sensor performance validation across varied environments. Both devices benefit from AIH LLC’s FDA certification consultation services, which help ensure compliance with QMSR’s risk management framework throughout their lifecycle.
These examples highlight how rigorous testing and documentation align with QMSR-defined risk classifications, paving the way for effective compliance strategies.
How to Ensure QSR Compliance in Wearable Devices
Design Controls and Risk Management
To establish reliable testing protocols, manufacturers must prioritize strong design controls and integrate risk management at every development stage. As of February 2, 2026, the FDA’s Quality Management System Regulation (QMSR) incorporates ISO 13485:2016, requiring manufacturers to align their quality systems with this international standard. This includes adopting a risk-based approach across all QMS processes, from supplier selection to software validation.
Maintaining a Design History File (DHF) is critical to demonstrate adherence to an approved design plan. For AI-driven wearables like aiSpine and aiRing, this means defining measurable design inputs that account for user needs, intended use, and environmental factors like electromagnetic compatibility. Software and firmware validation should align with the risk level of the device.
"The explicit integration of risk management throughout the clauses of ISO 13485 more explicitly establishes a requirement for risk management to occur throughout a QMS and should help industry develop more effective total product lifecycle risk management systems." – FDA Response to Comment 19
Risk management documentation should include a centralized Risk Management Framework (RMF) comprising a Risk Management Plan, Analysis Report, and Traceability Matrix. Tools like Failure Modes and Effects Analysis (FMEA) or Fault Tree Analysis (FTA) are essential for identifying hazards under both normal and fault conditions. For wearables monitoring vital signs or posture, conducting a production FMEA during design transfer can help identify manufacturing risks that might compromise sensor accuracy or data reliability.
Design validation must use initial production units or their equivalents to confirm the device meets user needs under actual or simulated conditions. It’s also important to document the transition from research to formal design and development, marking the point where full design controls are applied.
These practices ensure robust documentation and risk management, laying the groundwork for effective quality control and certification.
Quality Control and FDA Certification Support
Strong design controls provide the foundation for a streamlined quality system, which is crucial for certification. An ISO 13485:2016-aligned quality system is a key element of QSR compliance. While the FDA does not mandate ISO 13485 certification for U.S. market access, conducting a gap analysis can help manufacturers identify areas where their quality systems may need improvement. Embedding risk-based decision-making into processes like purchasing, production, and corrective actions is essential.
New manufacturers can benefit from consulting professionals to navigate the FDA certification process. For example, AIH LLC offers consultation services to assist wearable device makers in meeting the requirements for Class II and Class III devices. This support often includes guidance on maintaining FDA-specific records, such as ensuring complaint and service logs include the device’s Unique Device Identifier (UDI), as required under QMSR 820.35. Additionally, establishing formal design reviews at key milestones with independent reviewers is highly recommended.
"FDA expects medical device manufacturers, led by individuals with executive responsibilities, to embrace a culture of quality as a key component in ensuring the manufacture of safe and effective medical devices." – FDA Response to Comment 27
For AI-powered wearables, software validation should reflect the associated risk level. Devices like the aiRing, which monitor vital signs, require rigorous validation of their AI algorithms to ensure accurate performance across diverse user conditions. Manufacturers can utilize resources like the CDRH Learn platform, which offers over 200 educational modules to help with regulatory compliance. Additionally, marking proprietary documents as "confidential" before submitting them to the FDA can safeguard sensitive information under the Freedom of Information Act.
The design transfer process is another critical step, translating design outputs into production specifications within the Device Master Record (DMR). This ensures manufacturing processes consistently meet device requirements – an especially important consideration for wearables, where sensor placement, material selection, and assembly precision directly impact performance. Aligning the Corrective and Preventive Action (CAPA) system with risk management ensures that nonconformities are addressed in proportion to their severity.
Conclusion: QSR’s Role in Wearable Device Testing
The QMSR represents a transformative approach to testing and validating wearable medical devices. By aligning with ISO 13485:2016, it introduces a risk-based framework that spans the entire product lifecycle. For manufacturers of AI-driven wearables, this means embedding strict design controls, software validation, and process oversight into every stage of production.
At the heart of QSR compliance are design controls, which require manufacturers to maintain thorough Design History Files (DHFs), perform detailed risk assessments, and validate devices under real or simulated usage scenarios. The FDA also mandates objective validation of non-inspectable processes to ensure they consistently meet defined specifications.
A well-integrated CAPA system, paired with effective complaint handling, plays a critical role in resolving quality concerns systematically. These elements strengthen testing protocols by addressing deviations swiftly and effectively. When combined with detailed risk management documentation and traceability matrices, they form a solid foundation for meeting regulatory requirements and ensuring device reliability. Leveraging digital Quality Management Systems (eQMS) can further simplify documentation, automate processes, and maintain the audit trails needed for successful FDA inspections.
AIH LLC offers specialized consultation services to guide manufacturers through the FDA certification process for Class II and Class III devices. Their expertise includes software validation, risk management planning, and compliance documentation, addressing the heightened testing demands under QMSR. This support ensures that cutting-edge wearable devices like aiSpine and aiRing meet both regulatory expectations and user safety needs.
The QMSR’s alignment with international standards signals a move toward global consistency, reducing duplicate documentation while upholding stringent safety measures. This shift not only enhances regulatory efficiency but also reinforces consumer confidence in advanced health monitoring technologies.
FAQs
What does the shift from FDA QSR to QMSR mean for wearable medical device testing?
The FDA’s move from the Quality System Regulation (QSR) to the Quality Management System Regulation (QMSR) brings notable updates for wearable medical device testing. Manufacturers are now required to align their processes with international standards like ISO 13485:2016, which prioritizes streamlined and consistent quality management practices.
This transition places greater emphasis on management review records and demands stricter adherence to revised definitions and documentation requirements. The goal is to create more uniform and thorough testing protocols, enhancing the safety and performance of wearable medical devices.
What challenges do manufacturers face in meeting the updated QMSR requirements?
Manufacturers are grappling with major hurdles as they work to comply with the updated Quality Management System Regulation (QMSR). Key challenges include aligning their current quality management systems with ISO 13485:2016, addressing newly introduced regulatory demands, and ensuring all necessary adjustments are completed within the FDA’s two-year transition period.
This process typically involves extensive updates to documentation, comprehensive staff training, and overhauling operational workflows to meet stricter requirements. For wearable medical device manufacturers, the situation becomes even more complex as they navigate the integration of advanced technologies like AI into their products, adding another layer of difficulty to achieving compliance.
Why is risk management so important for wearable devices under the FDA’s QMSR framework?
Risk management plays a crucial role in the FDA’s Quality Management System Regulation (QMSR) framework. Its primary goal is to ensure that wearable devices remain safe and effective throughout their lifecycle. This involves a systematic approach to identifying potential risks, assessing their impact, and implementing measures to reduce or eliminate them.
For wearable medical devices, which often handle real-time health data, managing risks is even more critical. It not only protects patient safety but also ensures compliance with regulatory requirements. By addressing risks proactively, manufacturers can enhance confidence in their devices and minimize the chances of recalls or safety concerns.