
ISO 13485:2016 is now the FDA‘s primary quality standard for medical device manufacturing as of February 2026. This impacts spine health monitoring devices, which must meet strict safety and reliability benchmarks. Here’s what you need to know:
- Why It Matters: Spine monitoring devices, whether radiation-free or AI-powered, must comply with ISO 13485 to ensure patient safety and avoid costly recalls (up to $5 billion annually in the U.S.).
- Radiation-Free Devices: Focus on hardware reliability, sensor calibration, and material safety. Compliance involves maintaining detailed design and risk management documentation.
- AI-Powered Devices: Combine physical sensors with machine learning. They require additional software validation and risk management for algorithm updates, making compliance more complex.
- Key Challenges: AI devices face risks like algorithm drift and demand lifecycle-wide software validation under IEC 62304. Radiation-free devices must address physical design issues like sensor accuracy and biocompatibility.
Quick Takeaway: ISO 13485 compliance is essential for spine monitoring devices. While radiation-free systems focus on hardware, AI-powered devices must address both hardware and software complexities. Choose based on your priorities: simpler compliance or advanced data-driven capabilities.
Six steps to ISO 13485:2016 Certification and MDSAP Certification
sbb-itb-44aa802
1. Radiation-Free Spine Monitoring Devices
Radiation-free spine monitoring devices rely on physical sensors and mechanical components to function. These devices – such as wearable posture trackers and pressure-sensing mats – come with their own set of challenges when it comes to ISO 13485 compliance. The focus here is on ensuring hardware reliability, precise sensor calibration, and consistent manufacturing processes. By emphasizing radiation-free solutions, the importance of hardware compliance within the ISO 13485 framework becomes clear.
Compliance Documentation
Clause 4.2 of ISO 13485 requires maintaining a Medical Device File (MDF) that includes product specifications, intended use, labeling, and manufacturing procedures. Alongside this, a Design History File (DHF) must document all design changes, reviews, and approvals to establish a clear "evidence trail" for auditors.
When it comes to radiation-free devices, the documentation should include:
- Sensor specifications, such as angular accuracy of ±1°.
- Material biocompatibility certificates.
- Detailed assembly instructions.
This level of documentation creates a solid foundation for implementing risk management protocols.
Risk Management Protocols
Clause 7.1 highlights the importance of a risk-based approach, aligning with ISO 14971 standards. For radiation-free spine monitors, potential hazards include electric fields, leakage current, moving parts, and biocompatibility issues. To address these risks, follow a prioritized approach:
- Inherently safe design: Eliminate hazards through design choices.
- Protective measures: Integrate safety features directly into the device.
- Warnings and training: Provide clear safety instructions or user education.
The Risk Management File (RMF) should be continuously updated throughout the device’s life cycle, from initial design to decommissioning.
Validation and Verification
ISO 13485 also emphasizes the importance of design verification and validation to ensure the device meets both technical and user requirements.
- Design Verification (Clause 7.3.6): Confirms that sensor outputs match the specified design inputs. For example, verifying that a curvature sensor achieves its target accuracy of ±1°.
- Design Validation (Clause 7.3.7): Tests the device’s performance in real-world scenarios, such as clinical evaluations or usability studies.
- Process Validation (Clause 7.5.6): Applies to manufacturing steps that can’t be fully inspected afterward, like adhesive bonding or sterilization processes.
Waqas Imam from Advisera explains:
Validation shows the capability of industrial processes to attain planned results consistently.
To ensure no requirement is overlooked, create a traceability matrix. This links user needs to design outputs, verification tests, and validation results, providing a clear roadmap for compliance.
Post-Market Surveillance
Clause 8 outlines the need for procedures to gather feedback and handle complaints. For radiation-free spine monitors, this involves tracking real-world performance data to detect issues like sensor drift or material degradation. Key steps include:
- Collecting customer feedback and complaints.
- Using Corrective and Preventive Action (CAPA) data to update the risk profile.
It’s worth noting that failure to meet CAPA standards is the leading cause of FDA citations in the medical device industry. By maintaining continuous monitoring, manufacturers can ensure devices perform as intended over their lifecycle, reducing the risk of recalls. These hardware-focused measures set a high standard, especially when compared to AI-powered approaches discussed later in the article.
2. AI-Powered Spine Monitoring Devices
AI-powered spine monitoring devices bring a unique set of compliance challenges compared to radiation-free systems. Devices like the aiSpine posture monitoring system use physical sensors combined with machine learning to analyze spinal curvature and posture in real time. Achieving ISO 13485 compliance for these devices involves addressing both hardware reliability and software performance, requiring integration of standards like IEC 62304 (software lifecycle) and ISO 14971 (risk management).
As of February 2, 2026, the FDA has officially adopted ISO 13485:2016 as the primary quality system standard for medical devices in the U.S. by incorporating it into its Quality System Management Regulation.
Compliance Documentation
AI systems require additional software-centric documentation on top of standard hardware compliance. Clause 4.2 of ISO 13485 emphasizes the need for a Medical Device File (MDF), which for AI-powered devices includes software requirements specifications (SRS), architecture details, and verification evidence. For example, the aiSpine system must document how it converts sensor data into posture classifications and how alert thresholds are set. This ensures transparency, preventing the algorithm from being treated as a "black box" during audits.
Application Lifecycle Management (ALM) tools can simplify compliance by automating traceability, linking user needs to code changes and test results. This is especially helpful given that nearly half of life science companies (49%) still rely on paper-based systems, which often struggle with version control.
Risk Management Protocols
AI-powered devices face distinct risks compared to traditional hardware. While radiation-free systems deal with issues like sensor drift or material wear, AI devices must also address model drift – where algorithm accuracy declines as new data is introduced. ISO 13485 mandates a lifecycle-wide risk-based approach, with ISO 14971 serving as the guiding standard.
For AI spine monitors, the Risk Management File should identify potential hazards such as incorrect posture classifications, false alerts, or cybersecurity vulnerabilities. Clause 7.3 requires documenting decision logic using Explainable AI (XAI) models. Every algorithm update demands a formal risk reassessment to prevent new failure modes. Digital QMS platforms can streamline this process by automatically triggering risk reviews when AI parameters are updated.
These practices help AI-powered devices adapt to an ever-changing regulatory landscape.
Validation and Verification
Verification and validation ensure that AI spine monitors meet design requirements and perform effectively in real-world conditions. For these devices, this involves:
| ISO 13485 Clause | Process Type | Application to AI Spine Monitoring |
|---|---|---|
| 7.3.6 | Verification | Ensuring the algorithm accurately calculates spinal curvature angles from sensor data |
| 7.3.7 | Validation | Confirming the device effectively alerts users to poor posture, leading to better health outcomes |
| 7.5.6 | Software Validation | Ensuring consistent software performance across various mobile platforms or operating systems |
Design Verification confirms that the algorithm’s outputs match the design inputs, while Design Validation involves clinical studies or usability tests to prove the device works effectively in home or clinical settings. Clause 7.5.6 specifically requires validating software used in production or service before initial deployment and after any updates. Each algorithm update, therefore, triggers a new validation cycle.
A traceability matrix is crucial for connecting user needs to design inputs, verification tests, and validation results. Companies using AI-enabled QMS platforms have reported a 35% reduction in quality incidents within the first year by automating these connections.
After validation, continuous monitoring ensures that real-world performance aligns with laboratory results.
Post-Market Surveillance
Ongoing monitoring of algorithm performance is critical, particularly for metrics like false alerts and classification errors. Unlike hardware components that typically maintain steady performance, AI algorithms can evolve as they process new data patterns. This makes it essential to establish protocols for tracking algorithm accuracy over time.
Key surveillance activities include gathering real-world performance data, identifying unexpected classification errors, and using Corrective and Preventive Action (CAPA) data to update both the algorithm and risk profile. Customer feedback and post-market data must also feed into ongoing risk assessments to address unforeseen issues.
"Failure to meet CAPA standards is the number one trigger for FDA citations in the medical device industry." – Sumatha Kondabolu, Quality Expert
For devices like aiSpine, this involves monitoring metrics such as false positive rates for posture alerts, user adherence to recommendations, and overall algorithm performance across diverse populations. This feedback loop supports algorithm retraining and risk management updates, fostering continuous improvement throughout the device’s lifecycle.
Pros and Cons
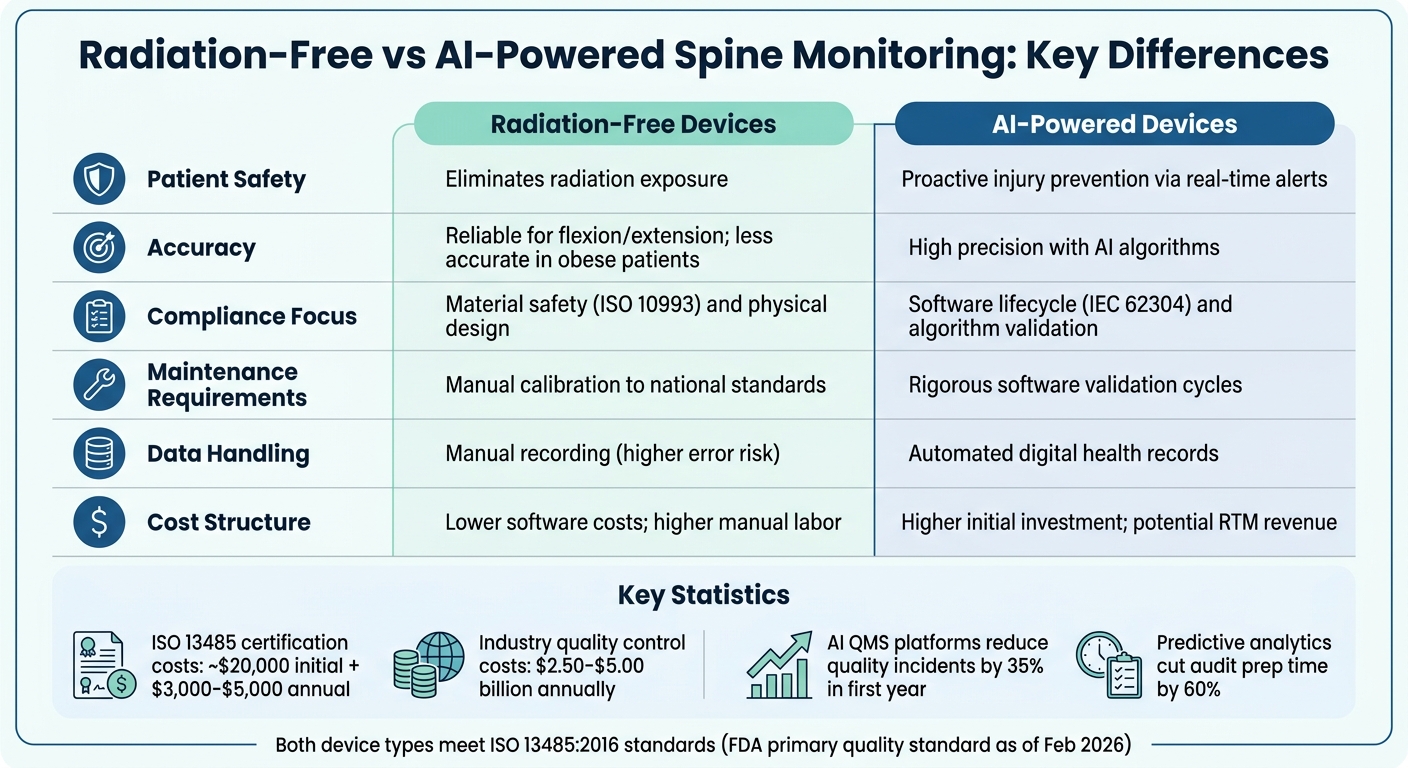
Radiation-Free vs AI-Powered Spine Monitoring Devices: ISO 13485 Compliance Comparison
Choosing between radiation-free and AI-powered spine monitoring devices involves weighing factors like compliance, accuracy, and costs. Both device types meet ISO 13485 standards, but they face unique challenges in quality management and risk control. Below is a breakdown of their respective benefits and limitations.
Radiation-free devices, such as SPIMON and Spinal Mouse, eliminate ionizing radiation, making them especially suitable for pediatric patients and individuals needing frequent monitoring. A 2023 University of Patras study with 40 healthy volunteers showed SPIMON measuring thoracic forward flexion at 71°, closely matching literature averages of 73°. However, these devices can struggle with accuracy in obese patients due to fat tissue movement obscuring actual spinal motion. On the compliance side, radiation-free devices focus on physical design controls and material biocompatibility (per ISO 10993), with simpler traceability requirements for components. Manual calibration to national standards is necessary, and failure to maintain calibration can lead to audit risks.
AI-powered devices, like aiSpine, use advanced algorithms and precision sensors to detect angular changes in real time, reducing errors often seen in manual assessments. These systems also enable Remote Therapeutic Monitoring (RTM), opening up new billable services for therapy adherence tracking. However, AI-powered devices come with stricter requirements, including IEC 62304 software validation and algorithm traceability. On the plus side, predictive analytics in these systems can cut audit preparation time by 60% and reduce quality incidents by 35% within the first year. The trade-off is a higher upfront investment and ongoing software validation costs.
Here’s a side-by-side comparison of key performance and compliance aspects:
| Criteria | Radiation-Free Devices | AI-Powered Devices |
|---|---|---|
| Patient Safety | Eliminates radiation exposure | Proactive injury prevention via real-time alerts |
| Accuracy | Reliable for flexion/extension; less accurate in obese patients | High precision with AI algorithms |
| Compliance Focus | Emphasis on material safety and physical design | Software lifecycle and algorithm validation |
| Maintenance | Requires manual calibration | Demands rigorous software validation |
| Data Handling | Manual recording with higher error risks | Automated digital health records |
| Cost Structure | Lower software costs; higher manual labor | Higher initial cost; potential RTM revenue |
Both types of devices aim to mitigate the $2.50 billion to $5.00 billion in annual costs linked to poor quality control in the medical device industry. The choice ultimately hinges on whether prioritizing radiation-free monitoring with simpler compliance or leveraging AI for continuous, precise monitoring better suits your needs.
Conclusion
ISO 13485 compliance plays a crucial role in ensuring patient safety and maintaining quality in spine health monitoring devices. Whether you’re working on radiation-free sensors or AI-driven wearables, this standard safeguards patients by enforcing stringent design controls, risk management, and traceability from a device’s inception to its retirement. Recent spikes in product recalls highlight the steep consequences of weak quality systems.
With the FDA’s Quality System Management Regulation (QMSR) now incorporating ISO 13485:2016, this standard has become the primary pathway for legal market access in both the U.S. and EU. It sets the stage for a unified framework that governs design, documentation, and ongoing quality management. Compliance is no longer a one-time milestone but an ongoing process requiring consistent documentation, automated traceability, and robust Corrective and Preventive Action (CAPA) systems. This is particularly critical for AI-powered devices, which must address challenges like algorithm drift and adhere to software lifecycle management under IEC 62304.
To stay ahead, manufacturers should focus on quality assurance early in the design phase rather than scrambling to fix issues post-market. Effective risk management and continuous documentation require streamlined systems. Transitioning from paper-based processes to electronic Quality Management Systems (eQMS) can simplify audit trails and document control. Additionally, conducting internal "dry runs" every three months helps ensure design, training, and purchasing records are audit-ready. For those developing AI-powered spine monitors like aiSpine, using explainable algorithms instead of "black-box" models is key, as it allows auditors to trace the logic from user requirements to code implementation.
"The companies that scale fastest aren’t the ones skipping documentation – they’re the ones systematizing it." – Orangesoft
The financial commitment for ISO 13485 certification includes an initial cost of about $20,000, annual fees ranging from $3,000 to $5,000, and daily audit charges of approximately $3,000. For the roughly 30,000 companies worldwide that have achieved this certification, it represents more than regulatory compliance. It signifies trust – trust from patients, clinicians, and regulators in an industry where safety is paramount.
FAQs
What changes for U.S. device makers after February 2026?
Starting February 2026, U.S. device manufacturers will need to comply with the updated Quality Management System Regulation (QMSR). This regulation integrates ISO 13485:2016, aligning FDA standards with international guidelines for medical device quality management systems. The goal is to ensure consistent product quality and regulatory compliance across the board.
Do AI spine monitors need IEC 62304 as well as ISO 13485?
When it comes to AI spine monitors, meeting specific regulatory standards is crucial. For quality management systems, ISO 13485 is often required. This standard ensures that the processes involved in designing and manufacturing the device meet stringent quality requirements.
For the software side of things, IEC 62304 is essential. It focuses on medical device software development, emphasizing software safety and lifecycle management. Depending on the monitor’s components and how its software operates, one or both of these standards may need to be followed.
What post-market data should spine monitors track to stay compliant?
Spine monitors need to keep an eye on post-market data, including complaints, user feedback, device performance, and reports of nonconforming products. Tracking this information not only ensures compliance with ISO 13485 standards but also plays a key role in driving ongoing quality improvements.